A part of our group is focused on the synthesis and study of ligands bearing imidazo[4,5-b]pyridine and imidazo[4,5-c]pyridine as the core heterocycles. This long-term project started with the development of a high-throughput synthetic approach leading to the mentioned imidazopyridine starting from immobilized amines, with 2,4-dichloro-3-nitroypridine and aldehydes as the joint building blocks (ACS Comb. Sci. 2014, 16, 558). We used this method for a rapid synthesis of the targeted chemical libraries of diversively substituted imidazopyridines for medicinal applications.
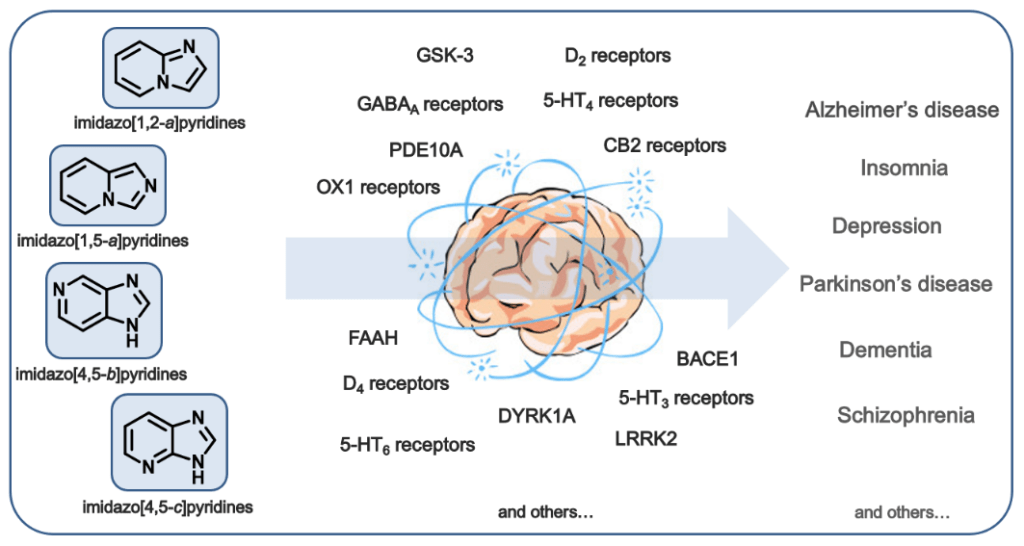
Imidazopyridines targeting the central nervous system (CNS)
Imidazopyridines act as potent ligands of diverse molecular targets localized in CNS, which represents the key idea of our research. Various imidazopyridines can be powerful modulators of diseases associated with CNS dysfunction such as Alzheimer’s disease, Parkinson’s disease, schizophrenia, depression, or sleeping disorders (Eur. J. Med. Chem. 2019, 181, 111569).
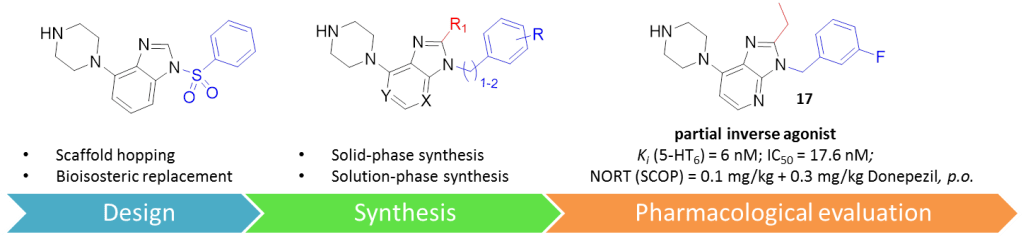
Taking this fact into account, we recently applied a scaffold-hopping approach to design novel N-benzyl imidazo[4,5-b] and imidazo[4,5-c]pyridine derivatives which have been identified as potent and selective 5-HT6R neutral antagonists (Eur. J. Med. Chem. 2018, 144, 716-729). We evaluated in vitro affinity of synthesized compounds for 5-HT6Rs, functional profile, and selectivity panel, followed by ADMET and pharmacokinetic properties for the most promising derivative. Finally, the pro-cognitive activity of the selected compound has been obtained in the novel object recognition (NOR) task in rats by measuring its ability to reverse drug-induced memory deficits.
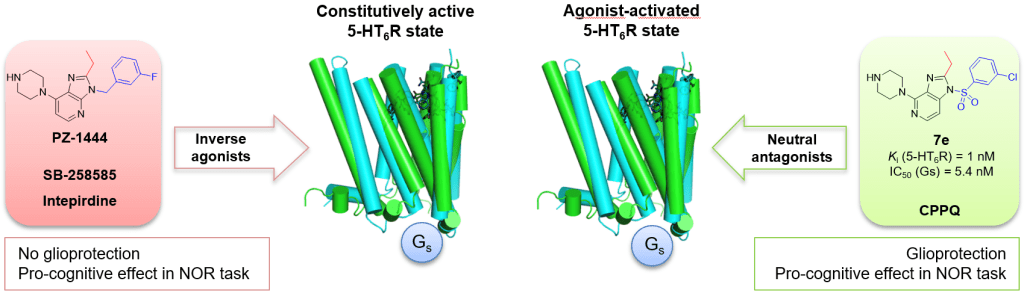
To further investigate the conformational effects of the 5-HT6 receptor, a focused library of N-benzenesulfonyl imidazo[4,5-b]pyridine and imidazo[4,5-c]pyridine derivatives were designed. The study identified compound 7e (1-[(3-chlorophenyl)sulfonyl]-2-ethyl-4-(piperazin-1-yl)-1H-imidazo[4,5-c]pyridine), which acts as a potent 5-HT6 receptor neutral antagonist at Gs, and does not impact neurite growth, in a process controlled by Cdk5 signaling pathways. Molecular dynamic simulations highlighted intramolecular constraints and receptor conformational changes. In cell-based assays, neutral antagonists (compound 7e and CPPQ), but not inverse agonists (SB-258585, intepirdine, and PZ-1444), displayed glyoprotective properties against 6-OHDA- and doxorubicin-induced cytotoxicity. Notably, both observed effects suggest that targeting the activated conformational state of 5-HT6R with neutral antagonists implicates the protecting properties of astrocytes. Additionally, compound 7e abolished scopolamine-induced memory deficits in the novel object recognition (NOR) task. In view of these findings, compound 7e is a molecular probe that can be used to further understand the functional outcomes of the different conformational states of the 5-HT6 receptor.
Except for ligands of 5-HT6R, our investigation of targeted libraries of imidazopyridines identified hit compounds that inhibited phosphodiesterase 4 (PDE4) with IC50 values comparable to rolipram and displayed different inhibitory potency against phosphodiesterase 7 (PDE7). The hit compound has shown beneficial effects in all studied animal models of inflammatory and autoimmune diseases (concanavalin A-induced hepatitis, lipopolysaccharide-induced endotoxemia, collagen-induced arthritis, and MOG35-55-induced encephalomyelitis). In addition, the compound showed a favorable pharmacokinetic profile after intraperitoneal administration; it was characterized by fast absorption from the peritoneal cavity and a relatively long terminal half-life in rats. It was found to penetrate the brain barrier in mice. The performed experiments shed light on the impact of PDE7A inhibition on the efficacy of PDE4 inhibitors in these disease conditions.
Imidazopyridines as the protein kinase inhibitors
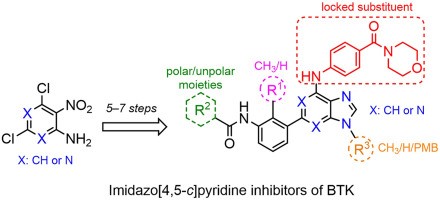
In comparison to other purine bioisosteres, imidazo[4,5-b]pyridine and imidazo[4,5-c]pyridine are relatively unexplored core heterocycles included in the ligands of protein-kinases. Inspired by this fact, we designed imidazopyridines as inhibitors of Bruton’s tyrosine kinase (BTK) (Eur. J. Med. Chem. 2021, 211, 113094). Two alternative synthetic routes for the simple preparation of desired compounds with variable substitutions at the N1, C4, C6 positions were introduced with readily available building blocks. Further, the developed synthetic approach was feasible for isomeric compounds bearing imidazo[4,5-b]pyridine scaffolds. In contrast to expectations based on previous studies, the imidazo[4,5-c]pyridine inhibitor exhibited significantly higher activity against BTK compared to its imidazo[4,5-b]pyridine isomer. An inherent SAR study in the series of imidazo[4,5-c]pyridine compounds revealed a remarkably high tolerance of C6 substitutions for both hydrophobic and hydrophilic substituents. Preliminary cellular experiments indicated selective BTK targeting in Burkitt lymphoma and mantle cell lymphoma cell lines. The inhibitors could thus serve as starting points for further development, eventually leading to BTK inhibitors that could be used after ibrutinib failure.
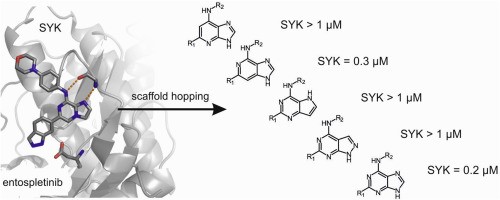
In the next study, we designed and prepared five isosteres in which the imidazo[1,2-a]pyrazine scaffold of entospletinib was altered to pyrazolo[3,4-d]pyrimidine, pyrrolo[3,2-d]pyrimidine, imidazo[4,5-b]pyridine, imidazo[4,5-c]pyridine and purine (Eur. J. Med. Chem. 2020, 204, 112636). The last two isosteres were the most potent SYK inhibitors, with IC50 values in the mid-nanomolar range. Importantly, three compounds also inhibited BTK more effectively than entospletinib. Further experiments then showed that BCR signaling was suppressed in Ramos cells by the potent compounds. Preliminary kinase inhibition screening also revealed LCK and SRC as additional targets. Our results further support the hypothesis that multikinase-targeting compounds could produce more robust responses in the treatment of B lymphoid neoplasms.